This
primary heart disease is due to disorders of sarcomeric proteins
of the heart muscle, showing a cellular disarray of myofibrils
(fibers of the heart muscle) on histologic, microscopic examination
( fig.
40b ).
This
cellular disarray leads to hypertrophy of the left ventricle
(increase thickness of the walls of the left ventricle), especially
the interventricular septum (IVS ), which is the muscular partition
between the right and left ventricles ( fig.
39a, fig.
39b, fig.
39c, fig.
39d, fig.
39f, fig.
39g, fig.
40a).
There
is also evidence to show that structural abnormalities of the
mitral valve are characteristic of many patients with this disease,
including increased size of the valve due to elongation and
abnormal insertion of the papillary muscle into the anterior
mitral leaflet ( figure
40c ).
In 25% of cases, the increase in size of IVS causes obstruction
of left ventricle (LV) outflow tract into the aorta (leading
to the disease called hypertrophic obstructive cardiomyopathy
(HOCM). The obstruction occurs when the anterior leaflet of
the mitral valve (MV) opposes the hypertrophic IVS through systolic
anterior motion (SAM), causing a pressure gradient between the
LV outflow tract and the area above the aortic valve ( fig.39i,
fig.40c,
fig.40d).
1. Genetically transmitted as autosomal dominant pattern
of inheritance in majority of cases.
2.
Remainder of cases: sporadic gene mutations.
Presentation
includes the following:
1.
Sudden death in 6% of children and young adults, and 1% in adults
45-60 years of age.
2. Symptoms include: dyspnea, paroxysmal nocturnal (sudden
shortness of breath at night), chest pain, presyncope, syncope
(fainting), fatigue, palpitations.
Physical
Examination reveals a harsh systolic murmur ( figure
39k ) best heard along the left sternal border of the chest
and the apex below the the left nipple. The most common form
includes diffuse hypertrophy of the IVS and anterolateral free
LV wall (70-75%) ( see: fig.
39a, fig.
39b, fig.
39f, fig.
39g, fig.
40a, fig.
40b, fig.
40f ).
Diagnostic
tests include:
1) EKG showing LV hypertrophy (LVH)
2) Holter monitoring may reveal the folowing abnormalities:
PVC's (extra or premature ectopic heart beats from the ventricles),
supraventricular tachycardia ( PSVT, see figure
2 and figure
3b,), sustained and nonsustained ventricular tachycardia
(VT, see: fig.6,
fig.7,
fig.8, fig.9a,
fig.9b,
fig.10,
fig.
11, fig.12,
fig.13,
fig.61)
and atrial fibrillation (see: figure
5a, figure 5b, figure
14 ).
3) Echocardiogram showing asymmetric IVS hypertrophy
( figure39i
), MR ( mitral regurgitation, figure
39j-a and figure
39j-b ) and increased pressure gradients between the LV
outflow tract and the ascending aorta by Doppler color studies,
often described as a dagger-shaped signal on continuous wave
Doppler studies (see figure
40a and figure
40f).
4) Exercise EKG testing in controlled circumstances.
5) Cardiac catherterization to verify pressure gradient
between the left ventricular outflow tract and the ascending
aorta.
6) magnetic resonance imaging (MRI)
7) tests for abnormal genes on chromosomes 1, 3, 11,
12, 14, 15, and 19
Hypertrophic cardiomyopathy accounts for 36% of deaths in athletes
younger than 35 (most common cause of sudden death in this age
group). Most sudden deaths are due to ventricular fibrillation
(VF) or ventricular tachycardia (VT) (see
fig. 61). These patients should avoid competitive sports
because of high risk of sudden death during physical exertion.
Electrophysiology
studies have been used to identify the above referred to cardiac
arrhythmias, which may lead to sudden death. In those with sustained
ventricular arrhythmias, placement of an automatic implantable
cardioverter defibrillator is justified (see
fig. 61).
Efficacy of placement in patients where syncopal episodes are
not caused by ventricular arrhythmia is less well established.
Betablockers, calcium channel blockers, and amiodarone may relieve
symptoms, but do not prevent sudden death.
Prophylaxis against infection of the heart valves with outflow
tract obstruction is indicated.
Nitrates
and ace inhibitors (heart medications) are contraindicated.
Surgical resection (myectomy) has been done in 5% of those with
IVS obstruction with gradients of 50mm or more.
Successful
surgical myectomy can significantly reduce or abolish the degree
of MR due to SAM without the requirement for concomitant mitral
valve surgery in all patients without independent mitral valve
disease. For patients without independent mitral valve disease,
there is a relationship between the LVOT gradient and MR jet
area (table
1, table
2,
fig. 39h below).
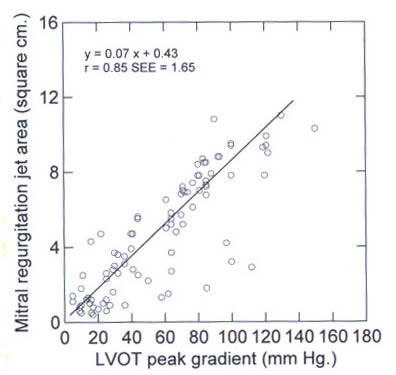
Figure 39h
The relationship
between the LVOT gradient and the mitral regurgitation jet area.
LVOT left ventricular outflow tract.
LVOT
= left ventricular outflow tract;MAC mitral annular calcification;
MRJA mitral regurgitation jet area; MV = mitral valve; MVP mitral
valve prolapse; PG peak gradient.
Values are expressed as mean ± SD.
<0.001 by analysis of variance and pairwise testing.
LVOT = left ventricular outflow tract, MR = mitral regurgitation.
(Yu,E.R.C.MD,and others,Mitral Regurgitation
in Hypertrophic Obstructive Cardiomyopahy:Relationship to Obstruction
and Relief With Myectomy,Journal of the American College of
Cardiology, Vol.36, No.7,2000, PP.2219-2224 ).
Also,
replacement of the mitral valve with a low profile prosthetic
valve to relieve obstruction may be indicated, especially with
concomitant mitral valve disease, but exposes patients to the
inherent risk of prosthetic valves and anticoagulant therapy.
Some of these patients may be relieved with only myectomy.
SURGICAL TREATMENT
Operation is regarded as the standard treatment for those HCM
patients with obstruction to left ventricular outflow under
basal conditions (gradient approximately 50 mmHg), and severe
drug-refractory symptoms. Therefore surgery is performed to
relieve incapacitating symptoms and subaortic obstruction by
normalizing the markedly increased systolic intraventricular
pressures. General agreement is lacking, however, as to whether
symptomatic patients with marked outflow gradients- which are
present solely or predominantly under provokable conditions
such as exercise or with maneuvers in the catheterization laboratory
(e.g., isoproterenol infusion, amyl nitrite inhalation, or Valsalva
maneuver) - are appropriate operative candidates.
Ventricular septal myotomy-myectomy (Morrow operation) ( Fig.
39l ) is the surgical procedure of choice; a small amount
of muscle is removed from the basal anterior septum (usually
about 2 to 6 g) through an aortotomy. However, mitral valve
replacement has been employed in selected patients when the
operative site for muscular resection in the basal anterior
portion of the septum is relatively thin (i.e., approximately
18 mm) or when the distribution of septal hypertrophy is atypical.
Occasionally, patients have outflow obstruction from a mechanism
other than mitral valve systolic anterior motion. For example,
anomalous papillary muscle insertion directly into anterior
mitral leaflet without the interposition of the chordae tendineae
( Fig.
39m ) producing muscular mid-ventricular obstruction should
always be contemplated prior to surgery, since the operative
strategy may require a more extensive myectomy or possibly mitral
valve replacement. Suture plication of the anterior mitral leaflet
(in combination with myotomy-myectomy) has also been introduced
in patients judged to have a greatly enlarged mitral valve,
so as to reduce the likelihood that mitral valve systolic anterior
motion will persist postoperatively.
Intraoperative 2D echocardiography is
an important guide to mapping the distribution and magnitude
of septal hypertrophy and determining how the muscle resection
should be tailored to the distribution of septal hypertrophy
in the individual patient to achieve the desired hemodynamic
result and avoid iatrogenic complications such as ventricular
septal defect. Transesophageal echocardiography may also be
useful in assessing morphologic and functional abnormalities
during surgery, particularly of the mitral valve.
Results from a number of North American and European centers
employing septal myotomy-myectomy over the past 40 years, in
about 2000 patients, have demonstrated salutary hemodynamic
as well as symptomatic effects. Operative mortality at the most
experienced centers has improved over the past several years
and is presently less than 1 to 2 percent. Older patients with
associated cardiac lesions, such as coronary artery disease
requiring bypass grafting, may be at greater operative risk.
Several important effects of operation have been defined in
patients with HCM. First, in more than 90 percent )f patients,
myotomy-myectomy (or mitral valve replacement) abolishes or
substantially reduces the basal subaortic gradient and mitral
valve systolic anterior motion without importantly compromising
left ventricular function; this consequence of surgery appears
to be permanent, with no evidence that the gradient recurs postoperatively
or that spontaneous growth of septal musculature recurs in the
area of the resection. Second, the reduction in left ventricular
systolic pressure is associated with a significant and persistent
improvement in symptoms and exercise capacity in 70 percent
of patients approximately 5 years after operation as well as
with a demonstrable increase in myocardial oxygen consumption
and improvement in lactate metabolism.
In a minority of patients, even after surgical relief of outflow
obstruction, symptoms may nevertheless return (presumably due
to persistently impaired left ventricular filling or ischemia,
atrial fibrillation, or conduction abnormalities), and premature
cardiac death can still ensue many years postoperatively. Traditionally,
surgery has not been recommended for asymptomatic (or mildly
symptomatic) patients with outflow obstruction since, in addition
to the operative risk, definitive evidence is lacking that prophylactic
relief of outflow obstruction prolongs survival, diminishes
risk for sudden death, or mediates the development of symptoms.
ALTERNATIVES TO SURGERY
Dual-Chamber Pacing
Although the septal myotomy-myectomy operation
is the first therapeutic option for severely limited patients
without obstructive HCM, perhaps the major limitation of surgery
is the restricted availability of surgeons with the necessary
experience to readily afford patients with low operative mortality
and a high expectation of hemodynamic and symptomatic success
with myotomy-myectomy. In addition, some patients are not ideal
surgical candidates, either due to advanced age, insufficient
personal motivation, or a limiting medical disability unrelated
to HCM. Therefore it is a reasonable aspiration to develop and
pursue alternatives to operation for this small but important
subgroup of patients. However, proper patient selection for
such procedures is a paramount consideration.
Over the past several years there has been some interest in
the application of permanent dual-chamber pacing, as an alternative
to operative intervention, for severely symptomatic patients
with obstructive HCM who are refractory to drug therapy. Observational
and uncontrolled studies have reported pacing to be associated
with reduction in outflow gradient and amelioration of symptoms
in many patients over relatively short time periods. However,
this reported symptomatic benefit has not been consistently
accompanied by improved exercise tolerance documented by objective
parameters (e.g., treadmill exercise duration and measured oxygen
consumption). Randomized, double-blind, crossover pacing studies
have shown that the subjectively perceived symptomatic improvement
reported by patients is largely due to a placebo effect. In
addition, the effect of pacing on outflow gradient and symptoms
is variable and reduction in obstruction is often much more
modest than that achieved with surgery.
Other laboratory catheterization studies
report dual-chamber pacing to have deleterious effects on left
ventricular systolic and diastolic function. For these reasons
and because the underlying HCM disease process and the risk
for sudden death do not appear to be altered by permanent dual-chamber
pacing, this potential treatment modality cannot be regarded
as a primary treatment for HCM .
However, there may well be a therapeutic role br certain subsets
of patients with this disease. In one randomized study, those
patients approximately 65 years old showed the most convincing
benefit from pacing.
Alcohol Septal Ablation
A second, recently introduced potential alternative
to surgery is alcohol septal ablation, in which about 2 ml of
alcohol is injected directly into the first septal perforator
coronary artery for the purpose of producing an MI, septal thinning,
and reduced mitral valve systolic anterior motion. This procedure
is intended to mimic the morphologic and functional consequences
of ventricular septal myotomy-myectomy. At present the septal
ablation technique is associated with a risk similar to that
of surgery but is capable of producing a substantial reduction
in the basal gradient. As yet, there is little objective substantiation
for the improvement in symptoms reported by many patients over
short-term follow-up. This is of particular importance in assessing
symptomatic and functional changes for a disease in which pathophysiology
is complex and symptoms are variable, often difficult to assess
by history, and subject to a placebo effect.
As is the case with pacing, alcohol ablation
should not be regarded as a primary treatment for the disease
or one capable of reducing the risk of sudden death. Indeed,
there is concern that this intervention could paradoxically
increase the future long-term risk for life-threatening ventricular
tachyarrhythmias and sudden death-a risk directly attributable
to the intramyocardial scar produced by alcohol ablation (which
is not present following myotomy-myectomy) in a patient population
that already harbors an arrhythmogenic subrate and often a particularly
long period of risk.
Maron,B.J.,HYPERTROPHIC CARDIOMYOPATHY,HURST'S
THE HEART,10th Ed.,PP.1967-1983.
The family should be screened for HCM.
Advances in Understanding Hypertrophic
Cardiomyopathy
SAIDI MOHIDDIN
LAMEH FANANAPAZIR
National Heart, Lung, and Blood Institute
The single most common cause of sudden death
in otherwise healthy young people, the disease is inherited
by at least one in 1,000 to one in 500 of the general population.
Nine genes are implicated; continued research can be expected
to uncover others, along with modifying factors that offer hope
of inducing regression. New interventions are being tested for
value in addressing symptoms and risk of death.
Everyone has read of the sudden, unforeseen
death of an apparently healthy young person--sometimes a well-known
athlete who collapses during a workout or game. The death is
often described in the press as "inexplicable." Hypertrophic
cardiomyopathy (HCM) is the likeliest explanation. It is in
fact the single most common cause of sudden death in otherwise
healthy young people.
HCM is a complex disease. However, the past
decade has brought significant advances in the understanding
of its molecular basis. Fundamentally, HCM is a genetic disease
with autosomal dominant inheritance occurring in at least one
in 1,000 to one in 500 of the general population. The disease
is often undiagnosed and would be even more prevalent were asymptomatic
cases routinely recognized. Usually, it develops during adolescence,
with progressive myocardial hypertrophy during the period of
rapid body growth, but it may be present in childhood or even
before birth. The hypertrophy, which affects predominantly the
left ventricle, is usually more marked than in any other cardiac
disease. It represents hypertrophy and hyperplasia of several
cell types, including cardiac myocytes, fibroblasts, and smooth
muscle cells, along with excessive collagen and matrix deposition
in the extracellular space. The normal parallel arrangement
of myocytes is often disturbed. Hypertrophy is often accompanied
by dynamic left-ventricular outflow obstruction, diastolic abnormalities,
and myocardial ischemia. Thus, patients may complain of dyspnea,
chest discomfort, light-headedness, presyncope, syncope, tiredness,
or palpitations, with symptoms often induced by exertion, dehydration,
or sudden changes in body posture, or following a large meal.
Progressive hypertrophy after age 20 is uncommon,
but initial diagnosis even in old age is not. In fact, clinical
findings may be unremarkable. Alternatively, the arterial pulse
may be bifid with a sharp upstroke, and on auscultation a third
or fourth heart sound and a systolic murmur (often intensified
by the Valsalva maneuver) may be heard. The electrocardiogram
is often abnormal. Diagnosis is made by echocardiography, following
identification of a hypertrophied left ventricle (in adults,
a left ventricular wall thickness of at least 13 mm; in athletes,
15 mm) inexplicable except by HCM. Left-ventricular obstruction
and mitral regurgitation may also be seen at echocardiography.
Evaluative methods such as exercise stress testing and Holter
monitoring may reveal myocardial ischemia or arrhythmias.
In all phenotypic respects, and in the risk
of sudden death, HCM can be extremely varied, even among patients
from a single family who share the same HCM-causing mutation.
There is also a striking genotypic variance, in that several
mutations in each of nine genes have thus far been shown to
cause HCM. These genes code for protein components of the myocardium's
contractile unit, the sarcomere. Collectively, they account
for perhaps no more than half of all cases, so several unidentified
genes may also be involved. Certain noncardiac clinical features
present in several families suggest that nonsarcomeric genes
may cause the disease. The emerging question is what the genetic
basis of HCM implies for diagnosis and management.
Mutations and Sarcomere Malfunction
A complete etiologic explanation for HCM would
consist of a causal chain beginning with a specific mutation
and ending with the various manifestations of the disease. Such
an account is beyond current understanding, but study of abnormalities
identified at the molecular level will greatly assist in determining
the mechanisms linking genetic cause and phenotypic effect.
Very generally, a mutant gene codes for a defective protein
with an altered biomolecular function that interferes with normal
cardiac physiology. The phenotypic consequences are a direct
or indirect result of this altered function. For the nine genes
known to cause HCM, the process originates in the sarcomere.
A myocyte is composed of sarcomeres arranged
in series and in parallel. In turn, each sarcomere consists
of a thick filament paralleling a thin filament (Figure 1 in
text or Fig. 40h on website). The thin filament is anchored
to a dense transverse band known as the Z-line. Interaction
between the filaments results in their relative motion, shortening
the sarcomere and creating a contractile force that acts at
the Z-line to cause contraction of the entire muscle cell. In
molecular terms, the thick filament is composed principally
of the polymerized tails of numerous beta-myosin heavy chains,
which are intertwined in pairs, while the thin filament consists
chiefly of polymerized actin, which forms from actin monomers
in a fashion resembling two stacks of coins gradually twisting
around each other.
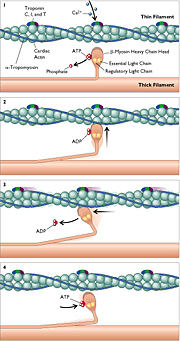 Figure.1
(Figure 40h)
Figure.1
(Figure 40h)
Click for enlargement
Interaction between myosin and actin is the
molecular basis of contraction. Each beta-myosin has a head
hinged to its long tail; in pairs, the heads extend away from
the heavy chain, oriented toward the thin filament, and are
therefore referred to as cross-bridges. Each head can bind actin
and the intracellular fuel ATP; the head also carries two proteins,
the essential and regulatory light chains of myosin, which are
thought to have a modulatory effect. The myosin-binding site
on actin is in the groove between the "stacks of coins."
In the resting state, the site is occupied by alpha-tropomyosin,
which inhibits actin-myosin interaction. Three other proteins,
troponins C, I, and T, are also components of the thin filament.
The muscle cell's electrical depolarization results in calcium
influx. Binding of calcium to troponin C induces a conformational
change in alpha-tropomyosin, exposing the myosin-binding site
on filamentous actin. The beta-myosin head can thus bind to
actin. The head can also hydrolyze its bound ATP, which dissociates,
causing a conformational change in the head that exerts directional
force on the thin filament. On binding the next ATP, the head
dissociates and beta-myosin returns to its former conformation.
If ATP and calcium remain available, the process can continue,
with progressive translocation of the thin filament relative
to the thick filament. Several other proteins have important
roles. Myosin-binding protein C is crucial for normal development
of the sarcomere and also regulates ATP hydrolysis by myosin.
Titin contributes to the passive elasticity of the myocyte and
may act as a sensor of the degree of myocyte stretch.
Within each of the nine genes known to cause
HCM--the genes encoding beta-myosin, the essential and regulatory
light chains, cardiac actin, alpha-tropomyosin, troponins C,
I, and T, and titin--several HCM-related mutations have been
identified. Most are missense mutations, permitting the biosynthesis
of a full-size protein with a substitute amino acid at one or
another position. For beta-myosin, almost all of the described
missense mutations are in the molecule's head or neck. The Arg403Gln
mutation is at the actin-binding site. It is a notably malignant
genetic defect, causing HCM in nearly all who have inherited
it, along with about 50% mortality by age 50 in the absence
of treatment. Other HCM-related beta-myosin mutations are in
other functional domains of the molecule, including the ATP-binding
region and the light-chain interfaces. Since HCM has an autosomal
dominant pattern of inheritance, HCM hearts have not only a
mutant gene but also a normal copy of the same gene. Incorporation
of the mutated protein into the heart's sarcomeres nonetheless
interferes with the normal function of the contractile apparatus.
This dominant negative gene effect contrasts with the carrier
status of autosomal recessive conditions, in which the normal
gene product can compensate for a missing or dysfunctional mutant
gene product.
It seems likely that, in one way or another,
all sarcomeric gene mutations associated with HCM impair the
function of the sarcomere as a molecular motor. This view is
supported by in vitro assays of the stiffness of normal and
HCM myocardial samples (a parameter proportional to the strength
of actin-myosin interaction), and by quantifying the ability
of beta-myosin to translocate actin. In the translocation test,
myosin is bound by its tail to a glass slide, leaving the head
free to interact with filamentous actin. For several different
mutations, translocation velocity is altered; in some cases,
such as the alpha-tropomyosin mutation Val95Ala, the velocity
is depressed, but for two essential light chain mutations, the
velocity is increased. This type of evidence, now obtained for
several mutations in several genes, supports the belief that
different mutations have distinctive functional consequences.
HCM-causing actin mutations have also provided
insight into the pathogenesis of cardiomyopathy. We have recently
identified five actin mutations in HCM patients. Three mutations
were de novo, present in patients but not in their parents,
and were therefore responsible for apparently sporadic HCM.
One was found in a 10-year-old with marked ST segment depression
and recurrent polymorphic ventricular tachycardia. Another two
actin mutations had already been found to cause a dilated cardiomyopathy
(DCM), in which left-ventricular walls are thinned and systolic
function deteriorates. Some 20% to 30% of DCM cases are hereditary,
with X-linked, autosomal dominant, and autosomal recessive inheritance
patterns all described. It is hypothesized that mutations interfering
with force generation between beta-myosin and actin cause HCM,
whereas those that interfere with force transmission from actin
to the Z-line result in DCM.
It should be added that several cardiac sarcomeric
genes--beta-myosin heavy chain, the essential and regulatory
light chains, and alpha-tropomyosin--are also expressed in skeletal
muscle. In some beta-myosin mutations, a skeletal myopathy develops
that resembles central core disease, in which mitochondria are
absent from the core of many slow muscle fibers. Light-chain
mutations have been associated with a skeletal myopathy involving
ragged red fibers similar to those seen in mitochondrial myopathies.
Accounting for Gross Changes
Several distinct heart morphologies have been
described in HCM (Figure 40g or Fig. 2 in text). These include
asymmetric septal hypertrophy (ASH), which affects mainly the
interventricular septum; reversed ASH, affecting the left ventricular
free wall; idiopathic hypertrophic subaortic stenosis (IHSS),
in which proximal septal hypertrophy obstructs left-ventricular
outflow; apical (also called Japanese) HCM; midcavity obstructive
HCM, in which hypertrophic papillary muscles abut in systole,
obstructing the midcavity of the left ventricle; and biventricular
hypertrophy, which may result in outflow obstruction. Although
patients sharing the same mutation may have markedly varying
phenotypes, genotype strongly influences morphology. For example,
midcavity HCM is frequently a result of mutations in the essential
or regulatory light chain (or the Leu908Val mutation of beta-myosin,
in the domain that binds light chains). An unusual apical meshwork
of myocardial tissue has been linked to a cardiac actin mutation.
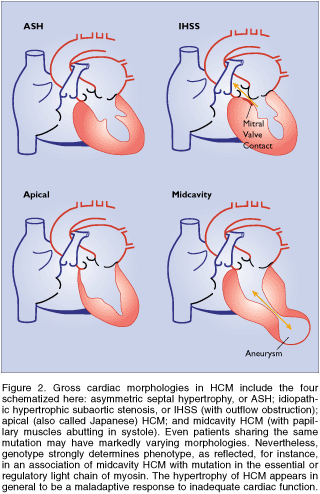
Fig. 40g (Fig. 2)
Currently, the most acceptable etiologic hypothesis
for HCM is that hypertrophy develops as a compensatory response
to sarcomeric dysfunction. The response is maladaptive, resulting
in the mature HCM phenotype. Myocytes are known to react to
excessive mechanical loads with a hypertrophic response involving
altered patterns of gene expression and the release of autocrine
growth factors such as angiotensin II. In hypertension, high
ventricular systolic pressures stimulate such a response. In
HCM, normal systolic pressures may be sensed by abnormal myocytes
as an excessive load, initiating an analogous hypertrophy. In
the short term, an increased left-ventricular wall thickness
and a reduced ventricular cavity size will lessen the magnitude
of wall tension required for systolic contraction. Indeed, most
HCM patients exhibit hyperdynamic left-ventricular contraction
and a supranormal ejection fraction. These appearances are deceptive;
ventricular systolic and diastolic volumes are smaller than
normal, and the high ejection fraction reflects not only lower
wall stresses but also reduced stroke volume.
Persistent stimuli to maintain hypertrophy
and promote maladaptive cardiac remodeling may arise from regional
differences in left-ventricular contractility and from myocardial
ischemia. In HCM, the causes of myocardial ischemia are incompletely
understood. But since the epicardial coronary vessels tend to
be enlarged and to exhibit rapid blood flow, it seems likely
that myocardial ischemia has more distal explanations. These
may include an excess oxygen demand by the hypertrophic, hyperdynamic
myocardium and abnormal intramyocardial arterioles narrowed
or blocked by endothelial and smooth muscle cell hyperplasia.
Eventually, myocardial necrosis and fibrosis may impair both
diastolic and systolic function and lead to cardiac failure
and arrhythmia.
Since the severity of left ventricular hypertrophy
often differs significantly among patients who share the same
HCM-related mutation--some family members with the mutation
may even remain unaffected--it appears likely that other genetic
factors modify the expression of the disease. Several possible
factors are under study, including polymorphisms in genes that
affect cellular hyperplasia or hypertrophy. Polymorphisms in
the gene for angiotensin-converting enzyme (ACE) are known to
affect the rate of production of angiotensin II. The clinical
significance of such variation--and the implications for therapy--remain
to be established. We are currently conducting a randomized,
double-blind, placebo-controlled study of the possible therapeutic
value of ACE or angiotensin II inhibition. Patients with nonobstructive
HCM receive ACE inhibitors, angiotensin II inhibitors, or placebo
and are examined at six months for changes in left-ventricular
hypertrophy, systolic and diastolic function, myocardial ischemia,
and electrophysiologic characteristics. Manipulation of factors
affecting expression of HCM-causing mutations may offer an opportunity
for intervening in the disease's development and prognosis.
Evaluation and Risk Stratification
Despite significant advances in the understanding
of the molecular basis of HCM and its management, risk evaluation
and prevention of sudden death in HCM remain limited. In efforts
to identify risk factors amenable to therapy, HCM patients have
been followed after clinical workup, Holter monitoring, exercise
myocardial scintigraphy, radionuclide angiography, hemodynamic
studies, and electrophysiologic evaluation. Several risk factors
have indeed been found, including a history of cardiac arrest
or syncope, a family history of sudden death of two or more
family members, sustained ventricular tachycardia induced during
electrophysiologic testing, particular genetic mutations, abnormal
blood pressure responses during exercise, and (in children)
myocardial ischemia.
In about 5% to 10% of HCM patients, myocardial
necrosis and fibrosis result in thinning of the hypertrophic
myocardium, leading to cardiac failure. These patients may be
at risk of sudden death. In almost 10% of HCM patients, atrial
fibrillation develops, with a risk of hemodynamic and thromboembolic
consequences (i.e., formation of atrial thrombi, with peripheral
or cerebral embolization). A poorly compliant left ventricle
has greater dependence on atrial systole for efficient diastolic
filling. During atrial fibrillation, a loss of atrial transport,
in combination with poor left-ventricular compliance, impairs
filling and depresses cardiac output, causing hypotension and
pulmonary edema. Lastly, about 20% to 30% of HCM patients exhibit
short runs of nonsustained monomorphic ventricular tachycardia
during one to three days of Holter monitoring. Ventricular arrhythmias
are frequent causes of cardiac arrest and sudden death. In particular,
nonsustained ventricular tachycardia may predict adverse outcome
in HCM patients with symptoms of impaired consciousness.
Overall, the findings suggest that asymptomatic
adults discovered to have HCM may not require risk evaluation
or intervention (Figure 40i), provided that the family has no
history of sudden death and the patient has no special need
for risk stratification, such as being an airplane pilot. Asymptomatic
children and adolescents may require more aggressive evaluation
and management, in view of the likelihood of rapid disease progression
and the risk of sudden death in adolescence. (The risk of HCM
sudden death during childhood is about 5% per year, compared
with an annual mortality of 1% to 2% in adults.) For children
and symptomatic adults, especially those with episodes of impaired
consciousness, workup may include 48-hour Holter monitoring,
exercise myocardial thallium scintigraphy, and cardiac catheterization
with electrophysiologic study (to evaluate left-ventricular
obstruction, arrhythmias, and their hemodynamic consequences)
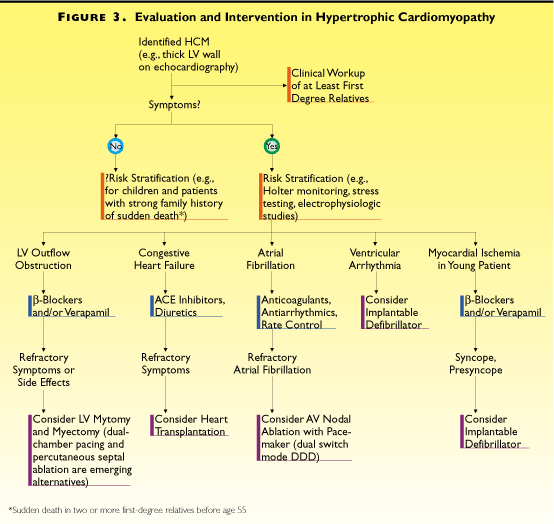
Figure 3:Evaluation and /intervention inHypertrophic
Cardiomyopathy
In collaboration with researchers at Johns
Hopkins University, we have recently evaluated the prognostic
value of an electrocardiographic parameter, QT-interval variability.
In all, 36 HCM patients with one or another of seven beta-myosin
mutations were compared with 26 age- and sex-matched healthy
controls. All subjects underwent Holter monitoring, from which
an algorithm identified the heart rate and QT interval for each
cardiac cycle. The mean QT interval and heart rate (and variance)
were similar in HCM patients and controls. Normally, the QT
interval is dynamically related to heart rate, so that at faster
heart rates the QT interval shortens. When this relation was
examined, HCM patients showed a significant decrease in the
association. The finding can also be described as an excessive
QT lability or a loosened coherence. QT lability was greatest,
and coherence with heart rate lowest, in patients with Arg403Gln,
the beta-myosin mutation associated with especially high mortality.
Studies had already shown that patients with HCM caused by Arg403Gln
are notably susceptible to myocardial ischemia and sudden death.
Clinical Implications of Genetic Discoveries
Since HCM is inherited, it becomes important
to examine the families of identified patients. First-degree
relatives (each of whom shares a 50% chance of having the HCM-associated
genetic defect) should be clinically evaluated by methods such
as echocardiography and electrocardiography, preferably at a
center familiar with HCM so that even mild manifestations are
recognized. For children, we suggest echocardiography at age
four or five, with repeat studies at four- or five-year intervals
as the child advances into and through adolescence. Following
diagnosis, it may be necessary to characterize the disease phenotype
more completely, for which invasive investigations may be indicated.
At this point, patients may benefit from referral to specialized
centers (such as our own).
Currently, genetic testing of patients' relatives,
and even of patients themselves, has logistical difficulties.
If the specific mutation responsible for HCM in a given family
is known, a test can easily detect its presence or absence in
individual family members. If the mutation is not known, each
of the nine genes known to cause HCM would have to be screened
for mutations. The process is laborious, has not yet been automated,
and has a detection rate of approximately 85%. Current methods
rely on the polymerase chain reaction to amplify each coding
region, or exon, of all nine genes, and on electrophoresis or
DNA sequencing to identify abnormalities. Currently identified
mutations, however, account for no more than half of all cases
of HCM, allowing less than a 50% likelihood of a positive finding.
Even then, clinical correlates with prognostic significance
have been determined for only a few of the known mutations.
A patient therefore has little chance of having a mutation whose
implications are fully described.
For genetic testing of hitherto unaffected
relatives of an HCM patient, further considerations arise. Not
all persons with an HCM genotype will have HCM. Indeed, in families
where the mutation has low penetrance, the cardiac phenotype
may skip one or more generations. Prenatal genetic diagnosis
may nonetheless force parents (one of whom may have experienced
considerable problems with HCM) to make difficult decisions.
Additionally, anxiety may follow the determination of a risk-related
genotype, and improper dissemination of such information may
affect employability and insurability. Counseling before testing
must include careful consideration of the risks as well as benefits
of genetic diagnosis.
For all the aforementioned reasons, genetic
diagnosis of HCM is not yet a routine component of clinical
assessment. However, the clinical correlates of specific mutations
will be appreciated only if genetic diagnoses are made more
frequently. Furthermore, the accuracy of screening family members
at risk of HCM is greatly enhanced if the causative mutation
is known. Recent advances in analytic methods such as DNA chip
technology are expected to facilitate large-scale mutation detection.
In HCM as in many other genetic diseases, the basic hurdle is
that any of a large number of different mutations in a large
number of different genes may be causative. A clinically applicable
mutation detection method must therefore be not only reliable
and rapid but also able to "interrogate" a large number
of DNA sequences.
DNA chips are silicon wafers with short DNA
segments (oligonucleotides) adsorbed onto their surface, which
thus becomes a grid of several thousand patches, each holding
a different oligonucleotide. The sequence of each oligonucleotide
is designed to be chemically complimentary to a sequence of
interest in human DNA. After incubation with human DNA preparations,
only oligonucleotides that encountered their complement become
double-stranded and are thereby detected. The chips are currently
used principally to quantify gene expression in one or another
tissue. Knowledge of patterns of gene expression in HCM hearts,
as compared with normal hearts, is expected to increase greatly
our understanding of mechanisms of hypertrophy and heart failure
and of responses to various therapies at a molecular level.
The knowledge may also suggest candidate genetic causes of HCM.
Furthermore, the chip technology lends itself to mutation detection.
If a chip's oligonucleotides are designed to be complimentary
to both normal and mutated gene sequences, the absence of normal
sequences and the presence of abnormal ones can be readily identified.
DNA chip technology may satisfy most requirements for clinical
applicability, but several practical difficulties remain to
be overcome, and rigorous testing must be conducted, before
this technique can be routinely used.
Technological issues aside, genetic studies
in HCM remain, for now, in the service of research. Efforts
to identify novel molecular causes of HCM rely principally on
two approaches. Candidate gene analysis requires the researcher
to hypothesize that a certain gene or class of genes may cause
HCM. The candidate genes are then screened for mutations in
HCM patients. By contrast, genetic linkage analysis is a statistical
approach to mutation detection. This method becomes usefully
powerful only in families where several members (usually more
than eight) are affected. The analysis takes advantage of several
hundred markers, each at a known chromosomal location. Informative
markers are spans of nucleic acid with a high frequency of variability
in any human population, allowing a given variant to be followed
through generations of a family. Markers are not causes of HCM,
but since all patients in a family affected by HCM will have
inherited the genetic cause from a common ancestor, the patients
are likely to have coinherited markers in close proximity to
the disease-causing gene. The positions of these markers identify
the approximate chromosomal location of the disease-causing
gene. This area can then be scrutinized in detail.
An important goal of our current efforts in
linkage analysis is to discover nonsarcomeric genes that cause
HCM--genes whose normal functions may be important clues for
understanding how the disease develops. To this end, we have
been analyzing large families with unusual disease presentations.
In one such family, HCM is accompanied by sensorineural hearing
impairment, in another by skeletal abnormalities that include
short metacarpals, and in a third by an accessory atrioventricular
conduction pathway (much as in Wolff-Parkinson-White syndrome).
Current and Novel Interventions
Management decisions (see Figure 40i; Fig.3
in text) are dominated by the need to address two problems:
disabling symptoms and risk of sudden death. In HCM, unlike
other cardiac disorders, the two are not well correlated. Patients
may have severe chest pain, shortness of breath, palpitations,
tiredness, or light-headedness but not die of HCM. On the other
hand, patients with no symptoms may die suddenly. Interventions
show the same dichotomy, in that they may address risk of death
without substantially relieving symptoms or relieve symptoms
without affecting risk of death.
Interventions that aim to improve prognosis
include implanted defibrillators for ventricular arrhythmias,
anticoagulants for atrial fibrillation, and ACE inhibitors (or,
failing that, heart transplantation) for cardiac failure. Interventions
addressing symptoms include a range of options for obstructive
HCM. Dynamic obstruction of left-ventricular outflow occurs
in about a third of HCM patients. Typically, the hyperdynamic
left ventricle and narrowed outflow tract increase the systolic
velocity of blood. The high velocity pulls a mitral valve leaflet
anteriorly, toward the interventricular septum, resulting in
outflow obstruction (and almost always mitral regurgitation).
First-line treatment is pharmacologic, with
negative inotropes (beta-blockers and verapamil), disopyramide,
and judicious doses of a diuretic for heart failure. If disabling
symptoms continue, patients have traditionally been referred
for cardiac surgery. Surgical options include myotomy and myectomy
(Morrow procedure) or mitral valve replacement. In myotomy and
myectomy, enough muscle must be removed from the septum to sufficiently
widen the outflow tract. Operative mortality depends on surgical
experience and is about 2% to 5%. Additionally, about 5% of
patients require a permanent pacemaker due to heart block. A
prosthetic mitral valve eliminates systolic anterior motion
and left-ventricular outflow obstruction, but the patient requires
life-long anticoagulant therapy and is subject to risk of prosthetic
malfunction.
Two novel nonsurgical alternatives are being
evaluated: dual-chamber pacing and percutaneous transluminal
septal ablation (PTSA). At the right ventricular apex, successful
pacing induces a paradoxic motion of the interventricular septum
away from the mitral valve, reducing left-ventricular obstruction.
The full benefits may appear gradually, so that after three
to six months of pacing, ventricular outflow pressure gradients
are reduced by an average of about 50%. Over longer periods,
cardiac hypertrophy may regress. Accurate pacemaker programming
and lead placement are critically important for optimal results.
In PTSA, a controlled mini-infarct is produced
in a target volume of septal myocardium. The interventional
cardiologist begins by identifying the septal perforator branch
(or branches) of the left anterior descending artery responsible
for perfusing the myocardial region where the anterior mitral
leaflet contacts the septum. After several precautions are taken,
this septal area is ablated by an infusion of ethanol through
an angioplasty catheter. Preliminary studies have reported improved
symptoms and a 70% reduction in ventricular pressure gradients.
The procedure is usually well tolerated but has been complicated
by conduction abnormalities requiring pacemaker implantation
and by ventricular arrhythmias and death. Long-term results
are not yet known. At the NIH, we are comparing dual-chamber
pacing with PTSA for patients with obstructive HCM and severe,
drug-refractory symptoms who otherwise would be candidates for
myotomy and myectomy. Thirty-five patients will be recruited
into each of the study's two arms (pacing or ablation), for
evaluation at six months. Important aspects of this study include
assessment of propensity to arrhythmia before and after therapy
and to cardiac remodeling following PTSA.
Conclusion
So far, nine HCM-related genes have been identified,
and in each of them several mutations have been described, for
a total of about 100 mutations. Although patients sharing the
same mutation may have markedly different clinical pictures,
genotype strongly determines disease expression--not only its
cardiac morphology but also its penetrance and prognosis. Even
the mechanism of sudden death appears to be strongly related
to genotype. To the extent that each mutation creates a variant
clinical picture, we therefore confront a variety of genetic
diseases whose clinical correlates, including outcome, have
not yet been thoroughly worked out. To further increase the
complexity, the genes found to date account for approximately
half of all HCM cases.
For the individual patient, it is currently
more important to evaluate the disease by its clinical manifestations
than by its genetic basis. Nevertheless, continued research
will not only improve our understanding of the function of the
known genes but can also be expected to uncover novel molecular
causes of familial cardiomyopathies, along with factors that
modify the resulting phenotype. Understanding these modifying
factors may offer our best hope of inducing regression. In the
interim, novel interventions are being tested for their value
in lessening symptoms and addressing the risk of death.